MC4R経路について
MC4R経路の機能/障害の原因
視床下部のはたらき
視床下部は、エネルギーバランス、食欲、体重の調節など、さまざまな神経内分泌機能を制御する脳の重要領域です1,2)。
視床下部は、下垂体の上方に位置する脳の鞍上部に存在する、相互に連結した神経核の集合体であり、以下を制御しています3,4)。
 睡眠、覚醒、概日リズム
睡眠、覚醒、概日リズム 疲労
疲労 体温調節
体温調節 口渇、塩分、水分のバランス
口渇、塩分、水分のバランス メラノコルチン4型受容体(MC4R)経路を介したエネルギーバランス
メラノコルチン4型受容体(MC4R)経路を介したエネルギーバランス
MC4R経路とは?
視床下部のメラノコルチン4型受容体(MC4R)経路は、食欲や食物(カロリー)摂取、エネルギー消費、さらに結果として体重を調節する重要なシグナル伝達経路です5-7)。
α-メラノサイト刺激ホルモン(α-MSH)は、MC4Rニューロンを活性化させることにより空腹感の軽減やエネルギー消費の増加を誘導します8-10)。食欲を適切に調節するには、MC4Rニューロンの活性化に十分なレベルのα-MSHが必要です。
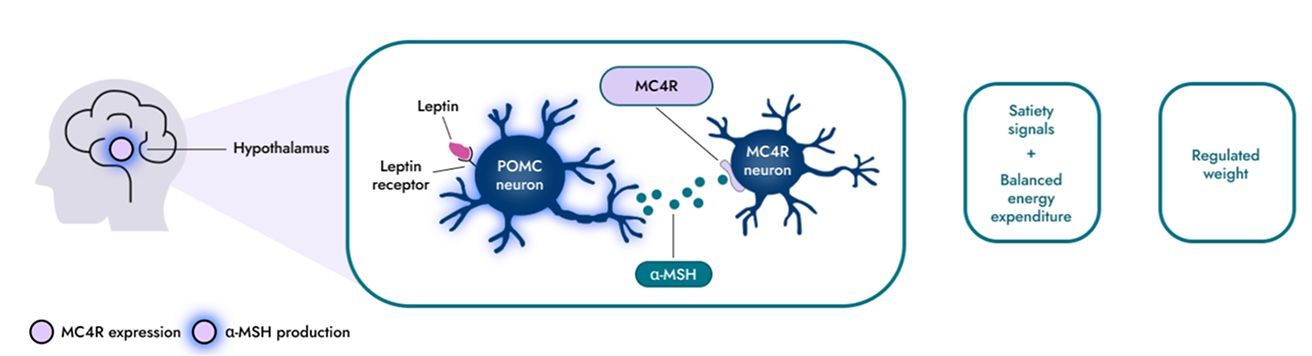
- 1
MC4R経路は、脂肪組織から放出されたレプチンに反応して、脳の視床下部領域内での神経活性化に関与することで、食欲を調節するシグナル伝達において重要な役割を果たします。
- 2
α-MSHはMC4Rに結合する神経ペプチドです。食欲を適切に調節するには、MC4Rニューロンの活性化に十分なレベルのα-MSHが必要です。
- 3
α-MSHによるMC4R活性化は、食欲や食物(カロリー)摂取、エネルギー消費、さらに結果として体重/体組成の調節に関与します。
MC4R経路障害とaHOの関係
MC4R経路の障害はα-MSH産生の減少につながる可能性があります。これによりMC4Rを介したシグナル伝達が障害され、過食(病的な、飽くなき食欲)やエネルギー消費の減少、およびその結果として急激かつ持続する体重増加につながります。後天性視床下部性肥満(aHO)患者では、これらの主要な臨床的特徴が観察されます11-13)。
後天性視床下部性肥満(aHO)の解説ページへ移動
後天性視床下部性肥満の病態
後天性視床下部性肥満(aHO)は、視床下部の物理的損傷または構造異常により生じ、MC4R経路の障害やその他の視床下部機能障害を伴う、急激かつ持続する体重増加を特徴とします14)。
MC4R経路の阻害は、α-メラノサイト刺激ホルモン(α-MSH)の産生を減少させ、過食やエネルギー消費の減少によるaHOの発症につながる可能性があります11-14)。
aHOの原因
最新情報を入手
各種資料のリリース情報やセミナー情報を受け取るには、サインアップが必要です。
現在準備中です
参考文献:
- 1.
Farooqi IS. Biol Psychiatry. 2022;91(10):856–59
- 2.
Yeo GSH, et al. Mol Metab. 2021;48:101206
- 3.
Van Santen HM, et al. Eur J Endocrinol. 2023;188:10.1093/ejendo/lvad009
- 4.
Waterson MJ, and Horvath TL. Cell Metab. 2015;22:962–970
- 5.
Krashes MJ, et al. Nat Neurosci. 2016;19(2):206–19
- 6.
Cone RD. Endocr Rev. 2006;27(7):736–49
- 7.
Loos RJF and Yeo GSH. Nat Rev Gens. 2022;23(2):120–133
- 8.
Van Santen HM. Horm Res Paediatr. 2025:10.1159/000543544
- 9.
Dimitri P, et al. Front Endocinol. 2022; 13:8468803
- 10.
Garnett MR, et al. Orphanet J Rare Dis. 2007;2:10.1186/1750-1172-2-18
- 11.
Roth CL, et al. Obesity (Silver Spring). 2015;23(6):1226–1233
- 12.
Hochberg I, et al. Obes Rev. 2010;11:709–721
- 13.
Roth CL, et al. Diabetes Obes Metab. 2024;26:34–45
- 14.
Madsen PJ, et al. J Neurosurg Pediatr. 2019;24(3):236–245
- 15.
Van Santen HM and Muller HL. Endocr Rev. 2025:10.1210/endrev/bnaf025
- 16.
Ruiz S et al. Eur J Endocrinol. 2022;186(6):R79–R92
- 17.
Maas AI, et al. Lancet Neurol. 2008;7(8):728–741
- 18.
Crenn P, et al. Clin Nutr. 2014;33(2):348–353
- 19.
Mele C, et al. Int J Mol Sci. 2021;22:10.3390/ijms22052686
- 20.
Jais A and Bruning JC. J Clin Invest. 2017;127:24–32
- 21.
Goszyonyi G, et al. Brain Structure and Function. 2020;225:1459–1482
- 22.
Etemadifar M, et al. Care Rep Med. 2012:10.1155/2012/768580
- 23.
Cerbone M, et al. E Clinical Medicine. 2020;19:100224
- 24.
Hietamäki J, et al. E Clinical Medicine. 2022;51:101556
- 25.
Beales PL, et al. Obes Rev. 2025;26:10.1111/obr.13915
- 26.
Baldini G and Phelan KD. J Endocrinol. 2019;241:R1–R33
- 27.
Roth et al. Obesity. 2011;19:36-42
- 28. Fonseca ACP, et al. J Diabetes Complications. 2017;31:1549–1561.
- 29.
Huvenne H, et al. Obes Facts. 2016;9:158–173
- 30. Hampl SE, et al. Pediatrics. 2023;151(2).
- 31. Malhotra S, et al. J Pediatr Genet. 2021;10:194–203.
- 32. Sivasubramanian R. and Malhotra S. Gastroenterol Clin N Am. 2023;52:323–332.
- 33. Argente J, et al. Lancet Diabetes Endocrinol. 2024;13(1):29–37.
- 34. Poitou C, et al. Eur J Endocrinol. 2020;183;R149–R166